Phylogenetic mean on a real tree¶
In this notebook we load a small primate phylogeny from a Newick string, seed the leaves with a trait value (brain volume, in cm³), and run the phylo_mean prebuilt to infer the trait at every internal node — the ancestral state reconstruction problem of comparative biology.
Along the way you’ll see:
hx.from_newick— Newick string / file →TreeHow BFS-ordered node indices relate to leaves named in the Newick source
The
phylo_meanprebuilt: each parent’s value = edge-length-weighted mean of its children, which is the closed-form estimator under a Brownian-motion model of trait evolutionA comparison against a naïve recursive average to see why edge-length weighting matters
hx.to_newick— round-trip back out to a Newick string
The maths is light; the focus is end-to-end workflow on a tree that looks like the ones you’d actually use.
import jax
import jax.numpy as jnp
import numpy as np
import matplotlib.pyplot as plt
import hyperiax as hx
from hyperiax.prebuilt import phylo_mean
def _dfs_x_positions(topo):
"""DFS left-to-right ordering of leaves → x ∈ [0, 1]."""
xs = np.zeros(topo.size)
next_leaf = [0]
n_leaves = int(topo.is_leaf.sum())
def visit(node):
if topo.is_leaf[node]:
xs[node] = next_leaf[0] / max(1, n_leaves - 1)
next_leaf[0] += 1
return xs[node]
# children of `node` in BFS order — ete3 BFS preserves Newick child order.
children = [i for i in range(1, topo.size) if int(topo.parents[i]) == int(node)]
cx = [visit(c) for c in children]
xs[node] = float(np.mean(cx))
return xs[node]
visit(0)
return xs
def plot_phylo_tree(topo, values=None, edge_lengths=None, ax=None, title=None,
cmap='viridis', vmin=None, vmax=None,
show_branch_labels=False, value_fmt='{:.0f}'):
"""Cladogram-style phylo tree drawing with leaf species names.
Uses cumulative edge length as y when ``edge_lengths`` is given
(proper time-tree), else falls back to ``-node_depth``. Leaf labels
are read from ``topo.names``.
"""
if ax is None:
_, ax = plt.subplots(figsize=(9, 5))
pos = np.zeros((topo.size, 2))
pos[:, 0] = _dfs_x_positions(topo)
if edge_lengths is not None:
age = np.zeros(topo.size)
for i in range(1, topo.size):
age[i] = age[int(topo.parents[i])] + float(edge_lengths[i])
pos[:, 1] = -age
else:
pos[:, 1] = -np.asarray(topo.node_depths)
# right-angled edges (cladogram style)
for i in range(1, topo.size):
p = int(topo.parents[i])
ax.plot([pos[p, 0], pos[i, 0], pos[i, 0]],
[pos[p, 1], pos[p, 1], pos[i, 1]],
'k-', lw=0.9, alpha=0.6, zorder=1)
if show_branch_labels and edge_lengths is not None:
mid_y = (pos[p, 1] + pos[i, 1]) / 2
ax.text(pos[i, 0] + 0.012, mid_y,
f'{float(edge_lengths[i]):.1f}', fontsize=7, color='gray',
ha='left', va='center')
# node markers
if values is None:
ax.scatter(pos[:, 0], pos[:, 1], s=260, c='lightsteelblue',
edgecolor='k', zorder=3)
else:
v = np.asarray(values)
sc = ax.scatter(pos[:, 0], pos[:, 1], s=320, c=v,
edgecolor='k', cmap=cmap, vmin=vmin, vmax=vmax, zorder=3)
plt.colorbar(sc, ax=ax, shrink=0.7, label='value')
# text overlays
names = topo.names if topo.names is not None else ('',) * topo.size
for i in range(topo.size):
if values is not None:
ax.text(pos[i, 0], pos[i, 1], value_fmt.format(float(np.asarray(values)[i])),
ha='center', va='center', fontsize=7, color='white', zorder=4)
if topo.is_leaf[i] and names[i]:
ax.annotate(names[i], (pos[i, 0], pos[i, 1]),
xytext=(0, -14), textcoords='offset points',
ha='center', va='top', fontsize=9, zorder=4)
if title:
ax.set_title(title, fontsize=11)
ax.set_axis_off()
return ax
1. Load a Newick tree¶
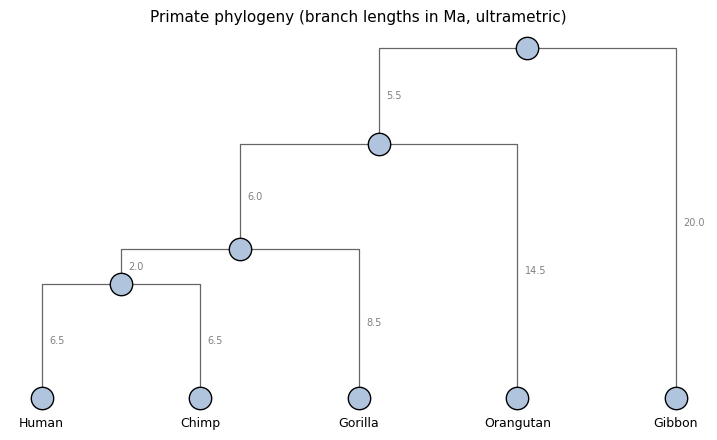
Our test tree is a small primate phylogeny — five great apes plus a gibbon — with branch lengths in millions of years (Ma), roughly consistent with the consensus divergence times on TimeTree.org:
Split |
Approx. age |
|---|---|
Human–Chimp |
6.5 Ma |
+Gorilla |
8.5 Ma |
+Orangutan |
14.5 Ma |
+Gibbon |
20 Ma |
Note that this is an ultrametric tree: every leaf sits 20 Ma below the root (the present is at the bottom of the tree).
PRIMATE_NEWICK = '((((Human:6.5,Chimp:6.5):2,Gorilla:8.5):6,Orangutan:14.5):5.5,Gibbon:20);'
tree = hx.from_newick(PRIMATE_NEWICK, schema={'estimated_value': ()})
print(tree)
print()
print(f"size: {tree.topology.size}")
print(f"depth: {tree.topology.depth}")
print(f"equal_degree: {tree.topology.equal_degree}")
print(f"leaves: {int(tree.topology.is_leaf.sum())}")
Tree(size=9, fields={edge_length: (), estimated_value: ()})
size: 9
depth: 4
equal_degree: True
leaves: 5
How Newick nodes map to BFS indices¶
The Newick parser produces a BFS-ordered topology — node 0 is the root, and parents[i] < i for i > 0. The names you wrote in the Newick string land on topo.names, and the colon-separated branch lengths land on tree.edge_length. Internal nodes have no name (empty string).
topo = tree.topology
print(f"{'idx':>3} {'name':<10} {'parent':>6} {'edge_len':>8} {'depth':>5} {'leaf?':>5}")
print('-' * 50)
for i in range(topo.size):
name = topo.names[i] or '(inner)'
leaf = 'yes' if topo.is_leaf[i] else ''
print(f"{i:>3} {name:<10} {int(topo.parents[i]):>6} {float(tree.edge_length[i]):>8.2f} {int(topo.node_depths[i]):>5} {leaf:>5}")
idx name parent edge_len depth leaf?
--------------------------------------------------
0 (inner) 0 0.00 0
1 (inner) 0 5.50 1
2 Gibbon 0 20.00 1 yes
3 (inner) 1 6.00 2
4 Orangutan 1 14.50 2 yes
5 (inner) 3 2.00 3
6 Gorilla 3 8.50 3 yes
7 Human 5 6.50 4 yes
8 Chimp 5 6.50 4 yes
plot_phylo_tree(topo, edge_lengths=tree.edge_length,
title='Primate phylogeny (branch lengths in Ma, ultrametric)',
show_branch_labels=True)
plt.show()
2. Seed leaves with a trait¶
For ancestral state reconstruction we need observed leaf values. We’ll use endocranial volume (cm³) — a continuous trait that’s easy to imagine and has a famous outlier (us).
Species |
Brain volume (cm³) |
|---|---|
Human |
1350 |
Chimp |
400 |
Gorilla |
500 |
Orangutan |
400 |
Gibbon |
100 |
The numbers are rough literature averages; the point is just that one species (us) is 3-4× the others.
BRAIN_VOL = {
'Human': 1350.0,
'Chimp': 400.0,
'Gorilla': 500.0,
'Orangutan': 400.0,
'Gibbon': 100.0,
}
# Look up each leaf's index from topo.names so we don't hard-code BFS order.
leaf_indices = np.where(topo.is_leaf)[0]
leaf_values = jnp.asarray(
[BRAIN_VOL[topo.names[i]] for i in leaf_indices], dtype=jnp.float32
)
tree = tree.at[topo.is_leaf].set(estimated_value=leaf_values)
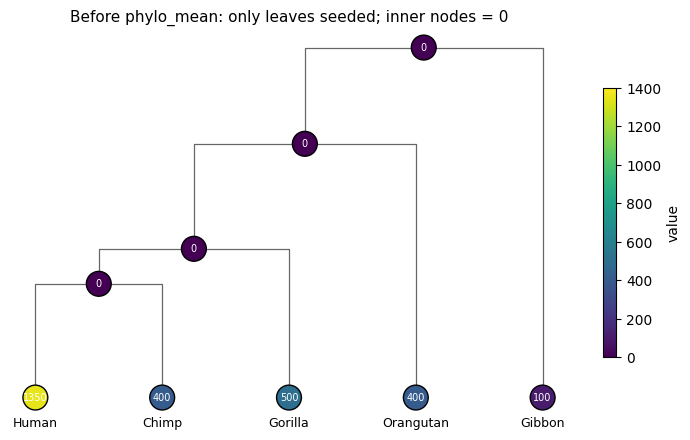
print('estimated_value (inner nodes are still zero):')
for i in range(topo.size):
print(f' node {i:>2} {topo.names[i] or "(inner)":<10} {float(tree.estimated_value[i]):>8.1f}')
estimated_value (inner nodes are still zero):
node 0 (inner) 0.0
node 1 (inner) 0.0
node 2 Gibbon 100.0
node 3 (inner) 0.0
node 4 Orangutan 400.0
node 5 (inner) 0.0
node 6 Gorilla 500.0
node 7 Human 1350.0
node 8 Chimp 400.0
fig, ax = plt.subplots(figsize=(9, 5))
plot_phylo_tree(topo, values=tree.estimated_value, edge_lengths=tree.edge_length,
ax=ax, vmin=0, vmax=1400,
title='Before phylo_mean: only leaves seeded; inner nodes = 0')
plt.show()
3. Run phylo_mean¶
The prebuilt sweep computes, at every parent:
where the sum is over the parent’s children \(c\) and \(\ell_c\) is the branch length connecting \(c\) to \(p\). Short edges → high weight (sister taxa pull the ancestor toward themselves); long edges → low weight (distant relatives contribute less). It is the maximum-likelihood estimator for the parent’s state under a Brownian-motion model of trait evolution along edges.
Implementation-wise it’s a single @hx.up sweep that reads children.estimated_value and children.edge_length and writes estimated_value back. We saw the explicit version in 02_writing_sweeps.ipynb; here we just import the prebuilt and call it.
sweep = phylo_mean()
inferred = sweep(tree)
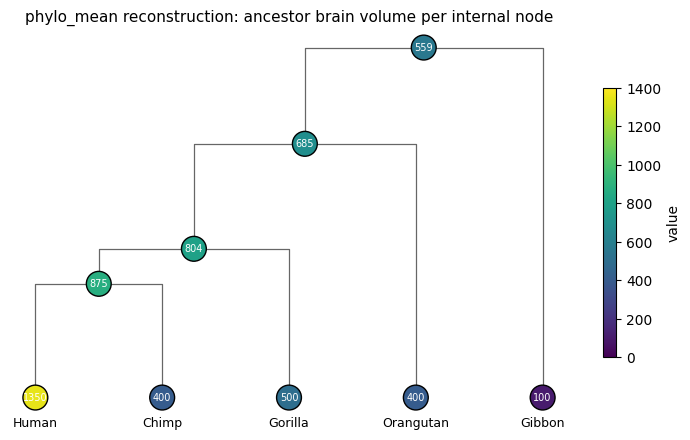
print('inferred values per node:')
for i in range(topo.size):
label = topo.names[i] or '(inner)'
print(f' node {i:>2} {label:<10} {float(inferred.estimated_value[i]):>8.1f}')
print()
print(f"Root (LCA of all 5 species) estimate: {float(inferred.estimated_value[0]):.1f} cm³")
inferred values per node:
node 0 (inner) 559.2
node 1 (inner) 685.5
node 2 Gibbon 100.0
node 3 (inner) 803.6
node 4 Orangutan 400.0
node 5 (inner) 875.0
node 6 Gorilla 500.0
node 7 Human 1350.0
node 8 Chimp 400.0
Root (LCA of all 5 species) estimate: 559.2 cm³
plot_phylo_tree(topo, values=inferred.estimated_value, edge_lengths=tree.edge_length,
vmin=0, vmax=1400,
title='phylo_mean reconstruction: ancestor brain volume per internal node')
plt.show()
4. Compare with a naïve recursive mean¶
How much did the edge-length weighting actually matter? Let’s run a baseline that uses children.value.mean(0) — every child contributes equally regardless of branch length — and compare the two reconstructions on the same tree.
@hx.up(reads_children=('estimated_value',), writes=('estimated_value',))
def naive_mean(node, children, params):
return {'estimated_value': children.estimated_value.mean(0)}
naive = naive_mean(tree)
flat = float(jnp.mean(leaf_values))
print(f"{'method':<20} {'root est. (cm³)':>15}")
print('-' * 38)
print(f"{'phylo_mean':<20} {float(inferred.estimated_value[0]):>15.1f}")
print(f"{'naive recursive':<20} {float(naive.estimated_value[0]):>15.1f}")
print(f"{'flat leaf mean':<20} {flat:>15.1f}")
method root est. (cm³)
--------------------------------------
phylo_mean 559.2
naive recursive 321.9
flat leaf mean 550.0
fig, axes = plt.subplots(1, 2, figsize=(15, 5))
plot_phylo_tree(topo, values=inferred.estimated_value, edge_lengths=tree.edge_length,
ax=axes[0], vmin=0, vmax=1400,
title='phylo_mean (edge-length-weighted)')
plot_phylo_tree(topo, values=naive.estimated_value, edge_lengths=tree.edge_length,
ax=axes[1], vmin=0, vmax=1400,
title='naïve recursive mean (children.mean(0))')
plt.show()
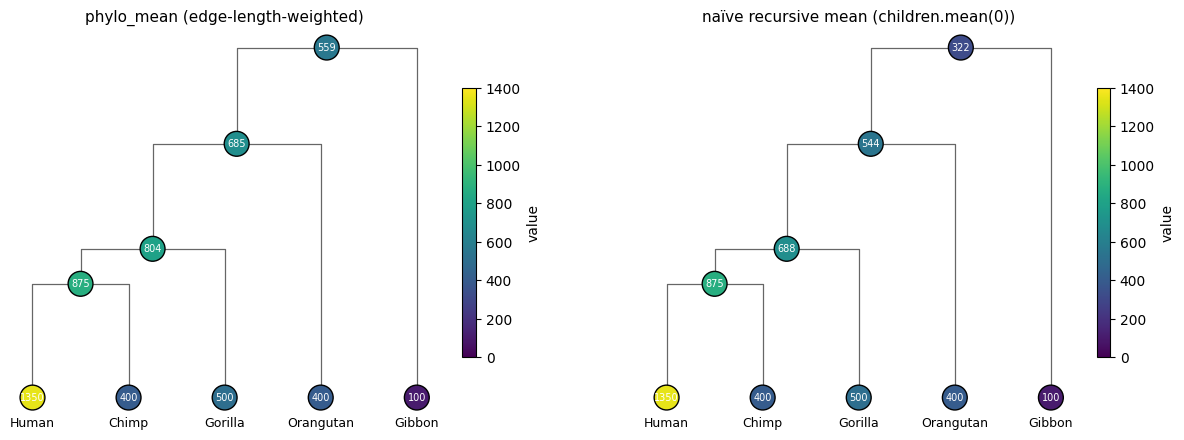
What you should see:
Naïve recursive mean is heavily biased by tree shape. At the root, it averages Gibbon (100) with the 4-ape subtree’s recursive mean — but the 4-ape subtree’s mean is already a recursive average that propagates Human’s outlier value through three levels of halving, so the value reaching the root from the apes side is much smaller than the flat leaf mean. The Gibbon side then drags it down further. Result is far below the flat leaf mean of 550.
phylo_meanis much closer to the flat leaf mean because the inverse-edge-length weighting compensates for tree imbalance. The 4-ape side has many short edges (high weight) and one long edge through Gibbon (low weight, 1/20 = 0.05). Gibbon contributes little; the apes contribute a lot — and the apes’ average pulls the root estimate up.
Neither is the “true” ancestral brain volume — that would require a full evolutionary model and is impossible to know without fossils. The point is that how you propagate up the tree matters, and phylo_mean is the structure-aware default.
5. Round-trip back to Newick¶
hx.to_newick converts the tree (including any updated edge_length) back to a Newick string. Internal node names that we never set come out as "". The inferred estimated_value field doesn’t appear in the Newick output — it isn’t part of the format.
round_tripped = hx.to_newick(inferred)
print('original: ', PRIMATE_NEWICK)
print('round-tripped:', round_tripped)
print()
print(f'identical? {round_tripped == PRIMATE_NEWICK}')
original: ((((Human:6.5,Chimp:6.5):2,Gorilla:8.5):6,Orangutan:14.5):5.5,Gibbon:20);
round-tripped: ((((Human:6.5,Chimp:6.5):2,Gorilla:8.5):6,Orangutan:14.5):5.5,Gibbon:20);
identical? True
Recap & next steps¶
What this notebook showed:
Loading Newick —
hx.from_newick(literal_or_path, schema={...})parses a string or file, returns aTreewithedge_lengthalready populated.BFS layout — the indices
0..N-1are a BFS traversal of the original Newick, and the names you wrote land ontopo.names. Look leaves up by name, don’t hard-code their indices.phylo_meanprebuilt — one-line ancestral state reconstruction under the Brownian-motion model.Comparing reconstructions — a custom
@hx.upsweep next to a prebuilt one is just two Python objects; you can swap them in the same pipeline.Round-trip writing —
hx.to_newick(tree)produces a Newick string you can save or paste into a tree viewer.
This was a point estimator: every ancestor got one number, no uncertainty. The natural next step is to ask “with what uncertainty?” — which leads to the Bayesian counterpart in notebook 04.
Where to go next:
04_phylo_bayesian.ipynb— full Bayesian inference on the same tree: closed-form posterior over ancestral states (Gaussian BFFG smoothing) with calibrated credible intervals.05_gaussian_bffg.ipynb— now infer the hyperparameters too, jointly with the latent states, via MCMC. BFFG inside the kernel.06_gaussian_nuts.ipynb— the same hyperparameter inference as point estimation: jax.grad-friendly building blocks compose with numpyro NUTS for full posterior inference.07_sde_bffg.ipynb— the research showcase: non-linear SDE bridges along edges, BFFG as an approximation, MCMC for full posterior.